
- Home/
- GATE MECHANICAL/
- GATE ME/
- Article
Structure and Properties of Engineering Materials Study Notes for Mechanical Engineering
By BYJU'S Exam Prep
Updated on: September 25th, 2023

Structure and Properties of Engineering Materials Study Notes- The existence of solids in nature occurs principally in two forms: crystalline and non-crystalline (amorphous), which differ substantially in their properties.
Crystals are solids that possess long-range order. There is a long-range order in crystalline solids, a regular pattern of arrangement of particles that repeats itself periodically can be seen over the whole crystal. Excluding the defects that can arise during the crystal growth, the arrangement of the atoms at one point in a crystal is identical to that in any other part of the crystal.
Table of content
The existence of solids in nature occurs principally in two forms: crystalline and non-crystalline (amorphous), which differ substantially in their properties.
Crystals are solids that possess long-range order. There is a long-range order in crystalline solids, a regular pattern of arrangement of particles that repeats itself periodically can be seen over the whole crystal. Excluding the defects that can arise during the crystal growth, the arrangement of the atoms at one point in a crystal is identical to that in any other part of the crystal.
Determination of the number of ways in which the component atoms are arranged in crystals and how the long-range order may be achieved is done using crystallography. Many physical and chemical properties are dependent on crystal structure and therefore the knowledge of crystallography is necessary if the properties of materials are to be understood and exploited.
Space Lattices- A space lattice provides the framework with reference to which a crystal structure can be described. An infinite array of points in three dimensions in which every point has a surrounding identical to that of every other point in the array is termed as a space lattice.
Basis– For obtaining a crystal structure, a group of atoms or an atom must be placed on each lattice point in a regular manner and such a group of atoms is termed as the basis which behaves like a building unit or a structural unit for the entire crystal structure.
Space lattice + basis → Crystal structure
UNIT CELL- A unit cell is the smallest component of the space lattice. The unit cell is the fundamental building block or structural unit of the crystal structure because of its geometry and atomic positions.
Lattice parameters of a unit cell- The unit cell is formed by primitives or intercepts a, b and c along X, Y and Z axes respectively. The three angles α, β and γ are called interfacial angles. One can easily determine the form and actual size of the unit cell if the values of these intercepts and interfacial angles are known.
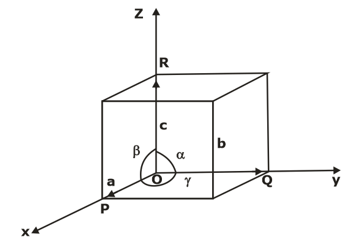 PRIMITIVE CELL- A geometrical shape which, when repeated indefinitely in 3-dimensions, will fill all space and is equivalent to one lattice point is termed as the primitive cell. All the primitive cells need not be unit cells but the reverse could be true.
PRIMITIVE CELL- A geometrical shape which, when repeated indefinitely in 3-dimensions, will fill all space and is equivalent to one lattice point is termed as the primitive cell. All the primitive cells need not be unit cells but the reverse could be true.
Types of crystals: Millions of tiny single crystals called grains constitute a crystalline solid and this is called microstructure and are said to be polycrystalline. The orientation of these grains is random with each other.
- Single crystals: Any single crystal, however, no matter how large, is a single grain.
- Single crystals of metals many cubic centimetres in the volume are relatively easy to prepare in the laboratory.
- Single crystals are produced artificially from their vapour or liquid state and they represent material in its ideal condition.
- Example sugar, sodium chloride (common salt), diamond, etc.
- Polycrystalline Crystals: An aggregate of many crystals usually known as polycrystalline separated by well-defined boundaries called grain boundaries.
- Due to the presence of with respect to each other and the obstruction of dislocations by the grain boundaries, a polycrystalline material is stronger than an ordinary one.
- Most of the materials exist in polycrystalline form
- As the polycrystalline materials exhibit the same properties in every plane and direction, they are called isotropic, single crystal is called anisotropic.
CRYSTAL FAMILIES AND CRYSTAL SYSTEMS– If all the atoms at the lattice points are identical, the lattice is said to be Bravais lattice. Crystals could be classified in terms of six crystal families, called anorthic, monoclinic, orthorhombic, tetragonal, hexagonal and iso-metric only upon careful measurement of the mineral specimen. A little expansion of this classification has been done by crystallographers. The crystal systems are sets of reference axes, which have a magnitude as well as a direction, and therefore are vectors.
|
Crystal family |
Crystal system |
Axial relationships |
Lattice type |
Examples |
|
Isometric |
Cubic |
a = b = c and |
P, F, C |
Au, NaCl, CaF2, CrCl, CaO (I) |
|
Tetragonal |
Tetragonal |
a = b ≠ c and |
P, C |
NiSO4, Sn, TiO3, and SnO2 |
|
Orthorhombic |
Orthorhombic |
a ≠ b ≠ c and |
P, B, F, C |
MgSO4, KNO3, and BaSO4 |
|
Monoclinic |
Monoclinic |
a ≠ b ≠ c and |
P, B |
2H2O, NaSO4, CaSO4, FeSO4 |
|
Anorthic |
Triclinic |
a ≠ b ≠ c and |
P |
K2Cr2O7, CuSO4, K2S2O8 |
|
Hexagonal |
Hexagonal |
a = b ≠ c and |
P |
AgCl2, Sio2, Zn and Graphite |
|
|
Rhombohedral |
a’ = b’ ≠ c’ and (Hexagonal axes) |
P or R |
SiO2, CaSO4, and CaCO3 |
Representation of symbols:
P → primitive,
B → base centred
C → body centred
F → face centred
Atomic radius- This is defined as half the distance between the nearest neighbours in the crystal structure of a pure Atomic radius is denoted by r and expressed in terms of the cube edge element a.
Atomic Packing Factor (APF)-The ratio of the total volume of all the atoms in one unit cell to the whole volume of the unit cell (also called relative density of packing (RDP)) is known as Atomic Packing Factor.
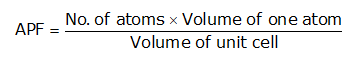 DENSITY OF CRYSTAL- This is defined as:
DENSITY OF CRYSTAL- This is defined as:
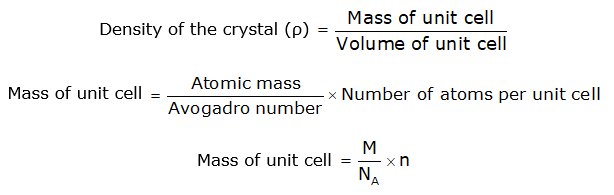
Where:
number of atoms in a unit cell and → n
Avogadro’s number → NA
the atomic weight, and →M
side of a cubic unit cell → a
![]() Linear density– The number of atoms per unit length along a specific crystal direction is termed linear density.
Linear density– The number of atoms per unit length along a specific crystal direction is termed linear density.
Planar density– The number of atoms per unit area on a crystal plane is termed Planar Density. This has significant effects on the rate of plastic deformation.
DIRECTIONS, LATTICE PLANES AND MILLER INDICES- There exists lattice and directions planes in a crystal that contain a large concentration of atoms. Different properties of crystals, particularly mechanical properties are related to the structure of the crystal through the help of crystal directions.
- Crystal Directions: To specify the direction of straight-line joining lattice points in a crystal lattice, we choose any lattice point on the line as the origin and the vector connecting this to any other lattice point on the line.
- Crystallographic Planes: The crystal lattice may be considered to make of an aggregate of a set of equidistant parallel planes, passing through the lattice points, which are known as lattice planes, or atomic planes.
- Miller indices of a direction are simply the vector components of the direction resolved along each of the coordinate’s axis, expressed as multiples of the unit cell parameters and reduced to their simplest form. They are denoted by [hkl].
- Miller indices of the plane a method to designate a set of parallel planes in a crystal by three’ numbers h, k and l usually written within brackets by (h k l).
- Interplanar spacings: Distances between different planes are shown by a number of parts of the body diagonal of a unit cell. The distance between planes can be calculated as per the miller indices as follows, (In a cubic system: a = b = c)
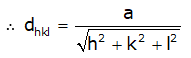 CO-ORDINATION NUMBER- The number of nearest atoms directly surrounding the particular atom. It is 6 for simple cubic, 8 for BCC and 12 for FCC. For closely packed structures, the value of this number is 12.
CO-ORDINATION NUMBER- The number of nearest atoms directly surrounding the particular atom. It is 6 for simple cubic, 8 for BCC and 12 for FCC. For closely packed structures, the value of this number is 12.
- For carbon atoms, the value of coordination number is 4, i.e. it has 4 nearest neighbours. The low coordination results in a comparatively less efficient packing of the carbon atoms in the crystal.
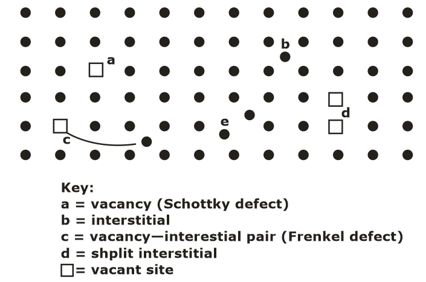
DEFECTS OR IMPERFECTIONS IN CRYSTALS- In ideal crystals atoms were arranged in a regular way. Certain defects or imperfections are always present in real crystals, and therefore, the arrangement of atoms in the volume of a crystal is far from being perfectly regular. Based on geometry, crystalline defects can be classified as :
- Point imperfections- The point imperfections, which are lattice errors at isolated lattice points, take place due to the imperfect packing of atoms during crystallization.
- Vacancies
- Interstitial Imperfections
- Frenkel defect
- Schottky Defect
- Substitutional Defect
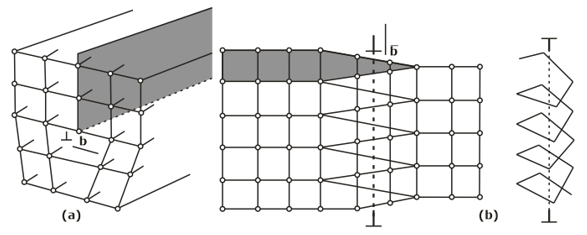
- Line imperfections- A dislocation is a linear disturbance (1 D) imperfection in a geometrical sense of the atomic arrangement occurring on the slip plane through the crystal.
- Edge Dislocation
- Screw Dislocation
- Climb Dislocation
- Surface and grain boundary imperfections
- Volume imperfections
Simple Cubic Cell (SCC)- In a SCC, there is an atom present at each corner of the cell and the centres of these atoms coincide with each corner.
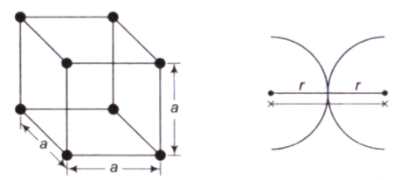
- (1/8)th part of the atom is present in the cell. The total number of atoms present in the crystal structure is 1
- r + r = a, Where r = Atomic radius, a = Lattice constant.
- Percentage APF = 52%
- Percentage of voids can be calculated as: 100 – 52 = 48%
Body-Centred Cubic (BCC) Structure- In BCC, An atom is present at the centre of the crystal and each corner coincides with a centre of an atom.
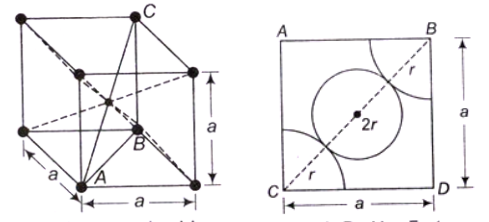
- Total effective number of atoms present in the crystal is 2
- Percentage APF = 68%
- Percentage of voids can be calculated as: 100 – 68 = 32%
Face Centred Crystal (FCC)- In FCC, each face has an atom and atoms are also present at the corners of the unit cell.
- Total effective number of atoms in the cell is 4
- Percentage APF = 74%
Percentage of voids can be calculated as: 100 – 74 = 26%
If you are preparing for ESE/ GATE or other PSU Exams (Mechanical Engineering), then avail Online Classroom Program for ESE and GATE ME:
Comprehensive Preparation for GATE & ESE ME Exams(500+ Hours of Live Classes, 300+ Quizzes for Practice and 60+ Mock Tests)
You can avail Test Series specially designed for all Mechanical Engineering Exams:
Get Unlimited Access to all 161+ Mock Tests
All the Best!
#DreamStriveSucceed.
Update BYJU’S Exam Prep, Best GATE exam app for Preparation


